Mechanisms regulating malignant haematopoiesis in AML
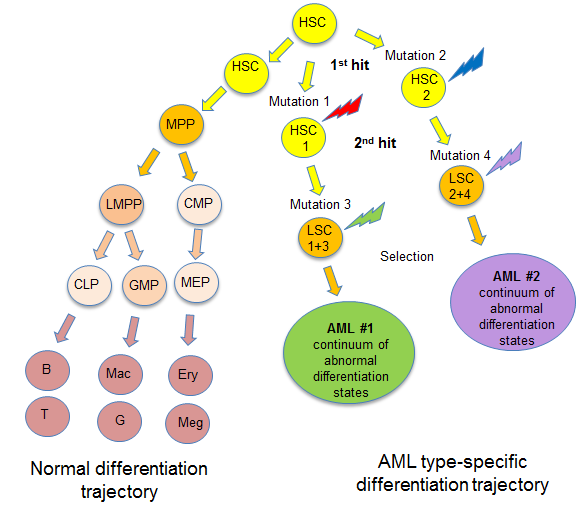
AML is a highly heterogeneous disease with many sub-types. Moreover, in individual patients with the same sub-type leukaemic cells undergo clonal evolution creating heterogeneous cell populations with different functional properties.
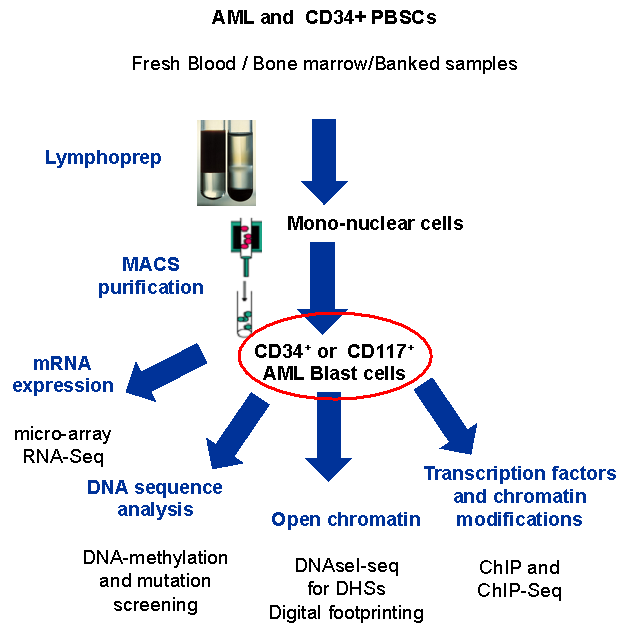
We focused on undifferentiated cells in karyotypically normal AML and only worked on samples where we could obtain a purified population of cells comprising at least 90% blasts. This involved purifying either the CD34+ or CD117+ population to get as close to the progenitor stage as possible.
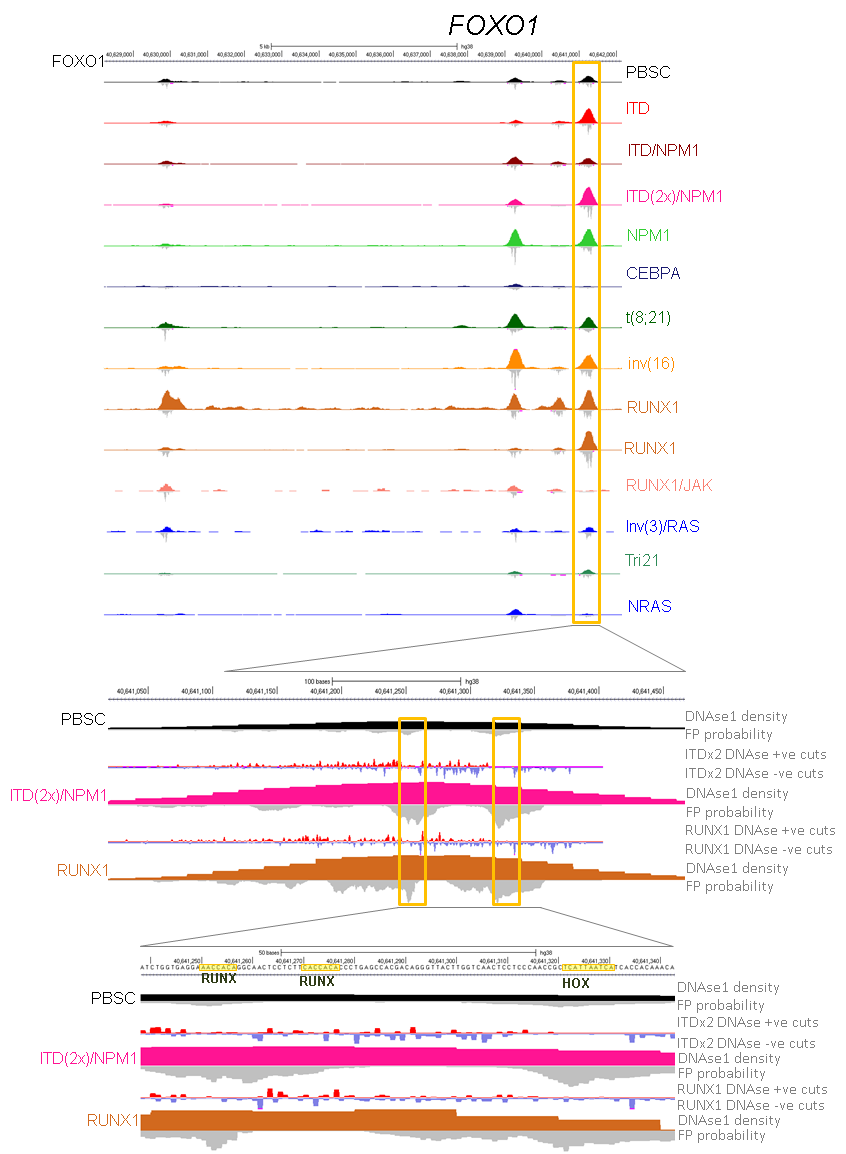
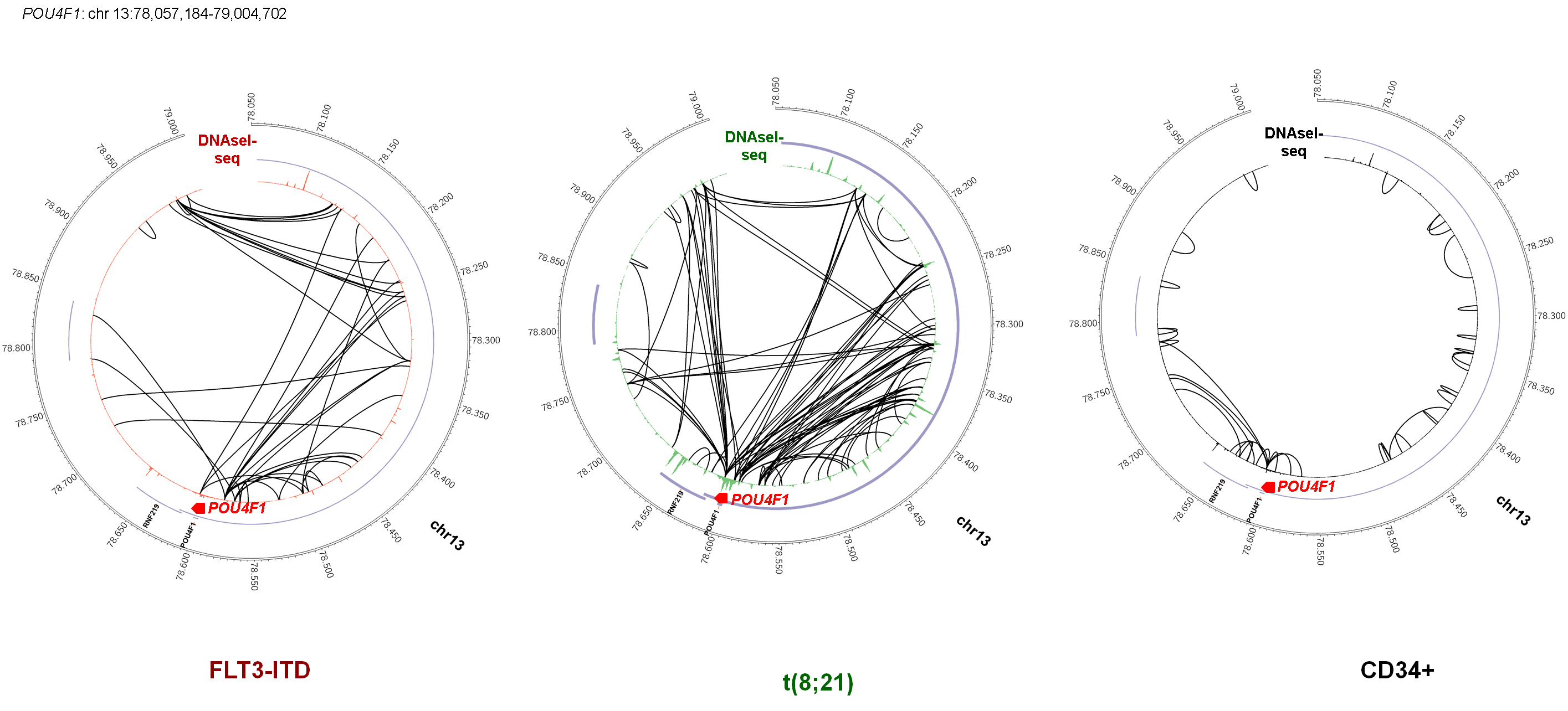
To be able to construct gene regulation networks associated with AML we performed a comprehensive set of genome-wide analyses summarised. This included (i) global DNaseI-Seq mapping of DNase I Hypersensitive site (DHS) patterns to identify aberrantly activated (and absent) cis-regulatory elements, (ii) identification of regions within DHSs protected from DNase I digestion (digital footprints) and the underlying specific motifs bound by TFs, (iii) ChIP-Seq assays to detect specific proteins bound at DHSs, (iv) global mRNA analysis to identify aberrantly regulated genes, and (vi) a DNA sequence screen for mutations within 55 candidate myeloid oncogenes and tumour suppressor genes to uncover pathways responsible for aberrant gene expression patterns